
ALS is a fatal motor neuron disease characterized by the degeneration of the upper and lower motor neurons. Most patients are diagnosed with sporadic ALS (SALS), while approximately only 10% of the patients are diagnosed with familial ALS (FALS). FUS gene mutations have been identified as a genetic cause of both SALS and FALS. FUS is an RNA/DNA-binding protein that mainly localizes to the nucleus and shares various similarities with TDP-43, which has been focused on as a causative protein of sporadic ALS. In the motor neurons of ALS-FUS patients, FUS is mislocalized to the cytoplasm and forms aggregates. Although previous animal experiments have suggested toxic gain-of-function, the pathophysiological mechanism is still unknown and key molecules for therapeutic targets have not been identified.
We identified DHX30, an ATP-dependent RNA helicase, as a FUS-interacting protein (Hikiami R et al., 2022). DHX30 localizes mainly within the mitochondrial matrix, and is crucial for mitochondrial ribosome assembly and translation. DHX30 gene mutations have also been reported to cause neurodevelopmental disorders. In the cultured cells and the spinal motor neurons of an ALS-FUS patient, DHX30 colocalized with FUS cytosolic aggregates. Furthermore, it was suggested that DHX30 may promote cytosolic aggregate formation.
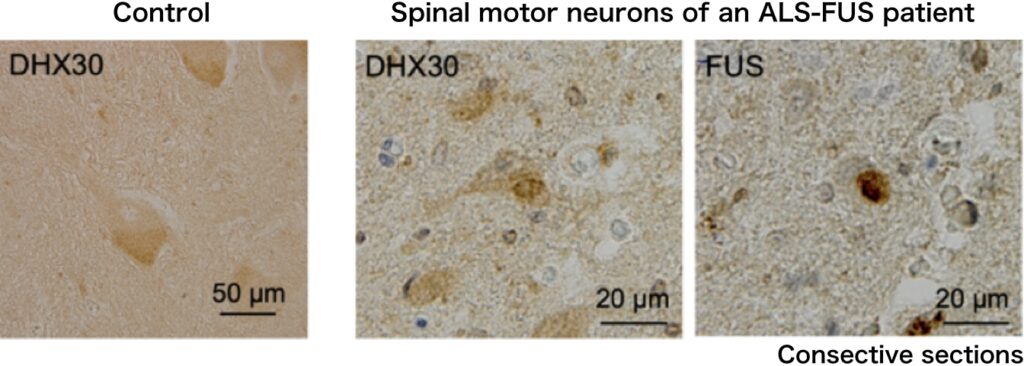
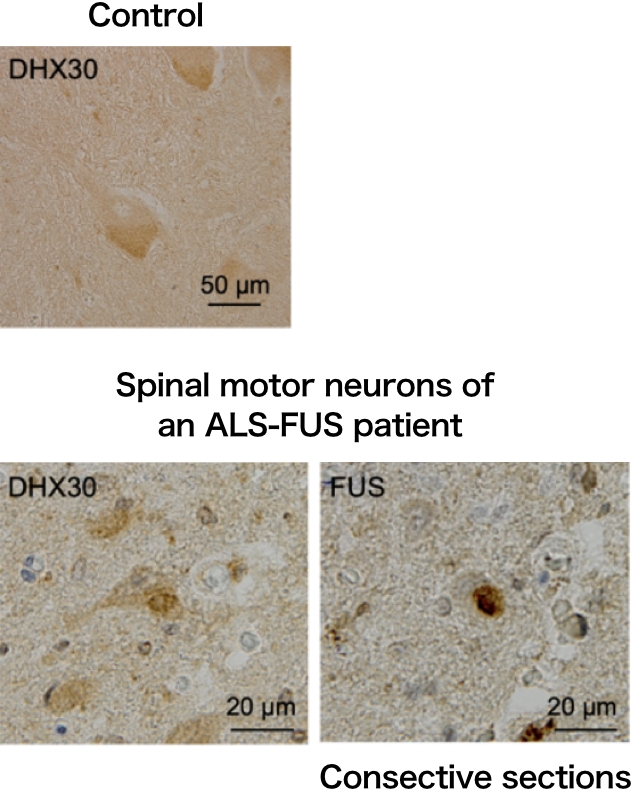
FUS mutants induced structural alterations of mitochondrial DHX30 mediated by aberrant disulfide bonds. The conformational defects resulted in decreased expression levels of the mitochondrially encoded proteins, OXPHOS assembly defect, and cytotoxicity, suggesting the loss of function of DHX30.
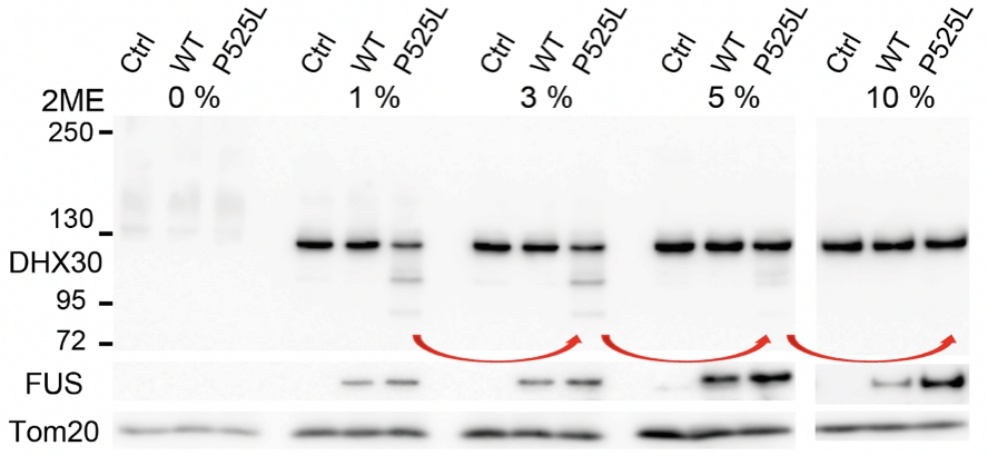
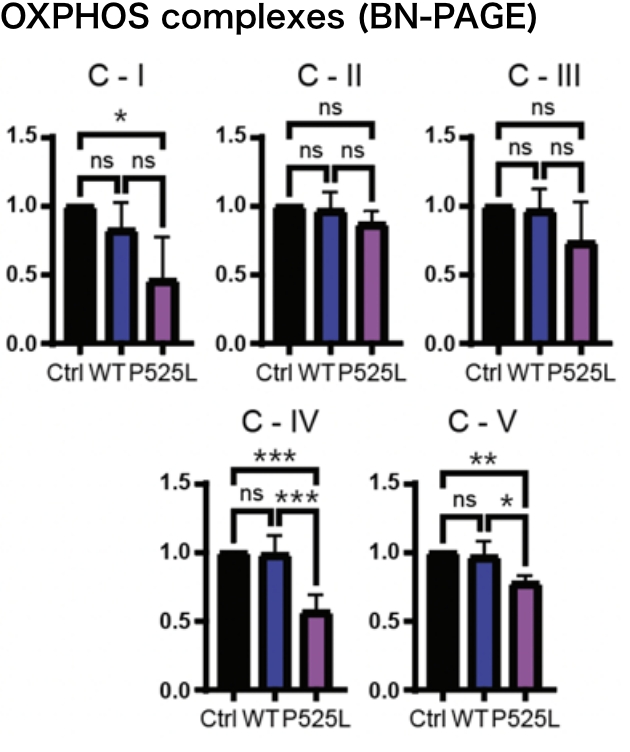
Based on the above, we believe that DHX30 is a potential therapeutic target in ALS-FUS and are trying to develop a therapeutic method.